β-amyloid (12-28) TFA 是 β-淀粉样蛋白 (β1-42) 的肽片段。 β1-42 是 42 个氨基酸的蛋白质,是老年斑核心的主要成分。β-amyloid (12-28) 具有聚集特性。β-amyloid (12-28) 有潜力用于阿尔茨海默氏病的研究。
编号:181342
CAS号:107015-83-8
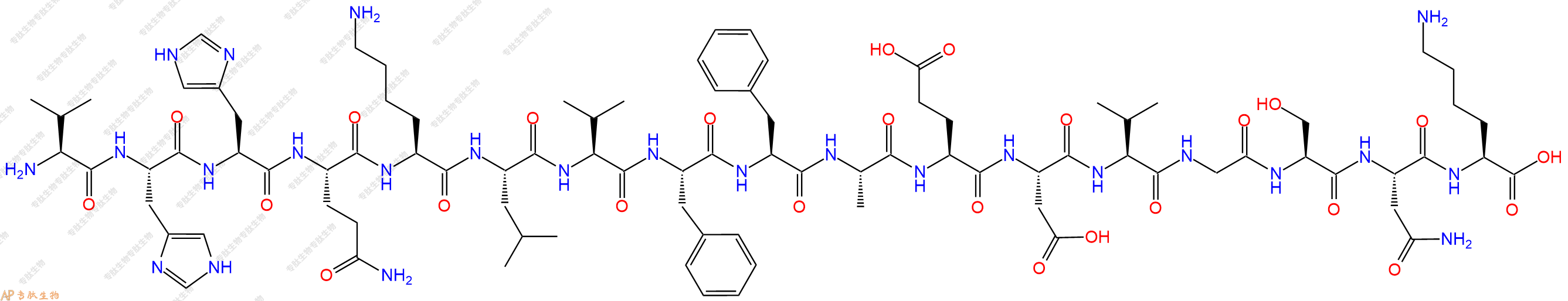
单字母:H2N-VHHQKLVFFAEDVGSNK-OH
| 编号: | 181342 |
| 中文名称: | 淀粉肽β-Amyloid (12-28)、Aβ12-28 |
| 英文名: | β-Amyloid (12-28) |
| 英文同义词: | Amyloid β-Protein (12-28) |
| CAS号: | 107015-83-8 |
| 单字母: | H2N-VHHQKLVFFAEDVGSNK-OH |
| 三字母: | H2N N端氨基 -Val缬氨酸 -His组氨酸 -His组氨酸 -Gln谷氨酰胺 -Lys赖氨酸 -Leu亮氨酸 -Val缬氨酸 -Phe苯丙氨酸 -Phe苯丙氨酸 -Ala丙氨酸 -Glu谷氨酸 -Asp天冬氨酸 -Val缬氨酸 -Gly甘氨酸 -Ser丝氨酸 -Asn天冬酰胺 -Lys赖氨酸 -OHC端羧基 |
| 氨基酸个数: | 17 |
| 分子式: | C89H135N25O25 |
| 平均分子量: | 1955.18 |
| 精确分子量: | 1954.01 |
| 等电点(PI): | 9.55 |
| pH=7.0时的净电荷数: | 1.45 |
| 平均亲水性: | -0.00625 |
| 疏水性值: | -0.33 |
| 消光系数: | - |
| 来源: | 人工化学合成,仅限科学研究使用,不得用于人体。 |
| 纯度: | 95%、98% |
| 盐体系: | 可选TFA、HAc、HCl或其它 |
| 生成周期: | 2-3周 |
| 储存条件: | 负80℃至负20℃ |
| 标签: | 阿尔兹海默症 |
β-amyloid (12-28) TFA 是 β-淀粉样蛋白 (β1-42) 的肽片段。 β1-42 是 42 个氨基酸的蛋白质,是老年斑核心的主要成分。β-amyloid (12-28) 具有聚集特性。β-amyloid (12-28) 有潜力用于阿尔茨海默氏病的研究。
β-amyloid (12-28) TFA is a peptide fragment of β-amyloid protein (β1-42). β1–42 a 42 amino acid protein , is the major component of senile plaque cores. β-amyloid (12-28) shows aggregation properties. β-amyloid (12-28) has the potential for Alzheimer’s disease research.
β淀粉样蛋白(Aβ或Abeta)是由淀粉样前体蛋白加工得到的多肽,含有36-43个氨基酸。众所周知,它是与阿尔茨海默病相关的淀粉样蛋白斑的成分,研究表明,Aβ是一个高度多功能的多肽,具有显著的非病理性活性。Aβ是在阿尔茨海默病患者的脑中发现的沉积物的主要成分。阿尔茨海默病患者脑中Aβ含量升高。 Aβ是脑实质和血管淀粉样蛋白的主要成分,它有助于脑血管病变和神经毒性。
Aβ(12-28)代表载脂蛋白E(apoE)的结合位点。ApoE是阿尔茨海默病的遗传危险因素,可促进有毒aβ的聚集。该序列包含一个疏水结构域(残基14-21)和一个β-转弯(残基22-28),将aβ14至21和29至40/42这两个疏水结构区彼此相对,允许aβ肽组装成原纤维。中性肽Aß(12-28)的二级结构主要由A-螺旋和无规卷曲组成。apoE与Aß残基12至28的相互作用不仅是非特异性疏水相互作用,而且在阿尔茨海默病(AD)的Aß病理机制中起着关键作用。Aß(11-28)和其他五个片段增强了全长Aß(1-40)的聚集。所有增强聚集的肽都含有残基17至20或30至35,表明这些区域对促进全长Aß聚集的重要性。
Aβ (12-28) represents a binding site for apolipoprotein E (apoE). ApoE is a genetically inherited risk factor for Alzheimer’s disease that promotes aggregation of toxic Aβ. This sequence encompasses a hydrophobic domain (residues 14–21) and a ß-turn (residues 22–28) which place two hydrophobic domains of Aß 14 to 21 and 29 to 40/42 opposite each other, allowing for the assembly of Aß peptides into fibrils. The secondary structure of Aß (12- 28), a neutral peptide, is dominated by a-helix and random coil. The interaction of apoE with residues 12 to 28 of Aß is not just a non-specific hydrophobic interaction but plays a pivotal role in the mechanism of Aß pathology in Alzheimer’s disease (AD). Aß (11-28) and five other fragments enhanced aggregation of full length Aß (1-40). All of the peptides that enhance aggregation contained either residues 17 to 20 or 30 to 35, indicating the importance of these regions for promoting aggregation of full-length Aß.

Extracellular amyloid-β peptide deposition into cerebellar plaques and formation of intracellular neurofibrillary fibers accompanied by the loss of neurons are characteristic histopathological lesions found in the brains of Alzheimer‘s disease patients. Individuals suffering from this disease show a gradual loss of cognitive functions and disturbances in behavior. Apart from some rare familial forms of the disease, the onset of Alzheimer‘s disease is usually above 60 years. Since the risk to develop the disease increases with age, Alzheimer‘s disease has turned into a major health and social problem in “first world” countries with an increasing proportion of older people, and is going to become one in emerging states. In this brochure we present amyloid peptides and related products for Alzheimer‘s disease research.
ALZHEIMER’S DISEASE
Alzheimer‘s disease (AD) is the prevalent cause of dementia in elderly people and has become one of the leading causes of death in developed countries together with cardiovascular disorders, cancer, and stroke. It is estimated that more than 46 millions of people suffer from AD all over the world. As age advances, the risk for developing AD increases. The frequency of AD at the age of 60-64 is about 1% and doubles approximately every five years. By the age of 90 and older, approximately 50% of the population suffers from this disease. AD is an irreversible and progressive neurodegenerative disorder. Symptoms include gradual loss of cognitive functions such as memory, verbal and visuospatial abilities, changes in personality, behavior, and activities of daily living. AD patients in the final stages are completely dependent on the care of others.
The characteristic lesions in the brains of AD patients were first described by the German neuropsychiatrist Alois Alzheimer in 1906 during the post-mortem examination of a mentally ill patient whose deterioration he had observed until her death. The lesions consisted of dense extracellular deposits, now designated as neuritic or senile plaques, and intracellular dense bundles of fibrils, which are now known as neurofibrillary tangles.
Currently, diagnosis of AD with adequate testing is approximately 90% accurate. It is based on the exclusion of a variety of diseases causing similar symptoms and a careful neurological and psychiatric examination, as well as neuropsychological testing. Imaging technologies for detecting amyloid plaques and tangles in vivo are becoming more precise and thus a valuable additional tool. Numerous potential biomarkers as α1 -antitrypsin, complement factor H, α2 -macroglobulin, apolipoprotein J, and apolipoprotein A-I for diagnosing AD are being evaluated. However, post-mortem histopathological examination of the brain is still the only definite diagnosis of this disease.
AD can be either inherited or sporadic. The inherited or familial AD is rare and comprises only 5-10% of all cases. Autosomal dominant mutations in the amyloid β/A4 protein precursor (APP) gene on chromosome 21 and the presenilin-1 or -2 genes on chromosomes 14 and 1, respectively, have been attributed to the early onset (before the age of 65) of this disease.
APP belongs to the type-1 integral membrane glycoproteins with at least 10 isoforms generated by alternative splicing of the 19 exons. The predominant transcripts are APP695, APP751, and APP770. A number of mutations within the APP gene have been detected in families with an inherited risk for early onset of AD. Usually, they are named after the region, in which they have been detected, e.g. the London APP717 mutations (V717I, V717F, V717G), the Swedish APP670/671 double mutation (K670N/M671L), the Flemish APP692 mutation (A692G), or the Dutch APP693 mutation (E693Q). The Swedish mutation of the β-secretase cleavage site of APP and mutations of positions 692-694 (Aβ 21-23), which strongly influence the aggregation behavior of Aβ, have been studied intensively.
A choice of relevant mutations in the Aβ region of APP is assembled in the table below.
| Exchanged Position in APP | Exchanged Position in Aβ | Designation |
|---|---|---|
| A673T | A2T | Icelandic |
| H677R | H6R | English |
| D678H | D7H | Taiwanese |
| D678N | D7N | Tottori |
| A692G | A21G | Flemish |
| E693D | E22∆ | Osaka |
| E693G | E22G | Arctic |
| E693Q | E22Q | Dutch |
| E693K | E22K | Italian |
| D694N | D23N | Iowa |
| L705V | L34V | Piedmont |
The presenilins are another group of proteins involved in the development of AD. Presenilins are integral membrane proteins with eight transmembrane domains localized in the endoplasmic reticulum and the Golgi apparatus. A multitude of mutations within the presenilin-1 and two within the presenilin-2 gene account for most of the cases of early onset of AD.
Genetic factors may contribute as well to the late onset of AD. Increased susceptibility is associated with the expression of different apolipoprotein E (ApoE) isoforms due to the polymorphism in the APOE gene on chromosome 19. In the central nervous system, ApoE has been implicated in growth and repair during development or after injury. Carriers of the APOEε4 allele show a higher risk in developing the disease than carriers of the other two possible alleles APOEε2 and APOEε3. The ApoEε4 effect seems to be dose-dependent since individuals with two of these alleles seem to be at two-fold higher risk to develop the disease than those with one allele. Polymorphisms of the α2 -macroglobulin gene on chromosome 12 and the gene coding low-density lipoprotein receptor-related protein 1 (LRP1), LRP1-C/T, have also been suggested to be a risk factor for the late onset of AD. However, further studies in this field are required.
A number of additional, most diverse risk factors have been proposed. These include gender, ethnic group, head trauma, cardiovascular diseases, and educational level.
AD THERAPEUTIC STRATEGIES RELY ON DETAILED KNOWLEDGE OF THE MOLECULES INVOLVED
Women, Hispanics, individuals who have experienced a head trauma earlier in life, and persons who suffer from cardiovascular diseases appear to have a higher risk of developing the disease.
The etiology of AD is still not completely understood. Initial research focused upon determining the molecular structure of the senile plaques and the neurofibrillary tangles originally described by Alois Alzheimer. The main constituents of the senile plaques were identified as cleavage products of APP, designated as amyloid β-peptides (Aβ peptides).
Depending on the composition and the fraction of fibrillar to non-fibrillar forms of these amyloid peptides, several kinds of senile plaques can be distinguished. Three types of proteases, α-secretase, β-secretase (or β-site APP-cleaving enzyme, BACE), and γ-secretase are involved in APP processing. APP can either be processed by the α- and γ- or by the β- and γ-secretases. The major two amyloid peptides identified in senile plaques, amyloid β-protein (1-40) (Aβ40) and amyloid β-protein (1-42) (Aβ42), are generated by successive proteolysis of APP by β- and γ-secretases. Cleavage of APP by β-secretase results in the release of the extracellular N-terminal protein fragment known as soluble APP-β molecule (sAPP-β). Then, the membrane-retained APP is further processed within the transmembrane domain by γ-secretase to yield either Aβ40 or Aβ42. The formation of Aβ40 and Aβ42 is a normal process, and both peptides can be detected in the plasma and cerebrospinal fluid (CSF) of healthy subjects.
In most studies, similar concentrations of Aβ40 have been measured in the CSF of both healthy controls and AD patients. On the other hand, Aβ42 concentrations in the CSF of AD patients are significantly lower than in normal controls, probably reflecting an increased deposition as insoluble plaques.
The neurofibrillary tangles found inside neurons of Alzheimer’s brains are composed of paired helical filaments whose main components are hyperphosphorylated forms of tau, a microtubule associated protein involved in promoting microtubule assembly and stabilization. Self-assembly into paired helical filaments is believed to be a result of hyperphosphorylation due to either the increased activity of protein kinases or the decreased activity of phosphatases.
Several lines of evidence support the view that the accumulation of Aβ42 in the brain is a primary event in the development of AD. Increased cerebral Aβ production appears to be characteristic for all the mutations within the APP and the presenilin genes of familial AD. In patients with Down syndrome (trisomy 21), elevated levels of APP and Aβ due to a third copy of the APP gene result in deposition of Aβ at an early age between 20 and 30.
Formation of neurofibrillary tangles is considered as a consequence of Aβ deposition with a further impact on the progression of the disease possibly due to disruption of axonal transport mechanisms in neurons.
The detailed knowledge about the molecules involved in AD has led to the development of several therapeutic strategies.

One strategy aims at the reduction of Aβ40 and Aβ42 by inhibition of either β- or γ-secretase activity or by clearance of Aβ in the brain by means of immunization with these peptides. Transition metals as Cu, Fe and Zn play an important role in the pathology of AD. Aggregation and neurotoxicity of Aβ are dependent on the presence of copper, so Cu-chelating agents showed promising effects in animal models. Another approach is the prevention of the cellular inflammatory response in the cerebral cortex elicited by the progressive accumulation of Aβ. Further preventive therapeutic strategies are based on the findings that cholesterol-lowering drugs such as statins and estrogen replacement therapy reduce the risk of developing AD. An additional treatment alternative would be the inhibition of the serine-threonine protein kinases, glycogen synthase kinase 3 (GSK3) and cyclin-dependent kinase 5 (CDK5), which are probably responsible for the phosphorylation of the tau protein. Inhibition of calpain, an enzyme showing increased activity in AD brains, led to promising results in animal studies. Calpain cleaves the CDK5 activator p35 leading to p25 formation and CDK5 overactivation.
Several acetylcholinesterase inhibitors such as tacrine, donepezil, rivastigmine, and galantamine have been approved for the treatment of mild to moderate AD by the FDA and other authorities. They act by reducing the deficits of the neurotransmitter acetylcholine associated with cognitive impairment in AD patients. The amantadine derivative memantine, an NMDA receptor antagonist, which was already used for the treatment of moderate to severe AD in Europe, has gained approval in the United States by the FDA as well.
A promising drug candidate, the β-secretase inhibitor verubecestat (MK-8931) developed for the management of mild to moderate AD, has moved to phase III. Moreover, the BACE inhibitor AZD3293 showed encouraging results in clinical studies. Antibodies as aducanumab and solanezumab, which have been designed to degrade plaques and lower the level of Aβ in the brain, have reached advanced stages of clinical testing for mild cases of AD.
Despite the many promising therapeutic approaches, AD still remains a major burden for the patients, their relatives, and the society.
| DOI | 名称 | |
|---|---|---|
| 10.1002/psc.418 | Structural, kinetic and cytotoxicity aspects of 12-28 beta-amyloid protein fragment: a reappraisal | 下载 |
| 10.1038/1565 | "Genetic dissection of Alzheimers disease and related dementias: amyloid and its relationship to tau" | 下载 |
| 10.1080/13506120701814723 | Potent anti-angiogenic motifs within the Alzheimer beta-amyloid peptide | 下载 |