|
多肽合成技术
多肽化学已经走过了一百多年的光辉历程,1902年,Emil Fischer首先开始关注多肽合成,由于当时在多肽合成方面的知识太少,进展也相当缓慢当时合成采用了苯甲酰,乙酰保护,脱去相当困难,而且容易导致肽链断裂。直到1932年,Max Bergmann等人开始使用苄氧羰基(Z)来保护α-氨基,该保护基可以在催化氢化或氢溴酸的条件下定量脱除,多肽合成才开始有了一定的发展。到了20世纪50年代,随着越来越多的生物活性多肽的发现,大大推动了有机化学家们对多肽合成方法以及保护基的研究,因此这一阶段的研究成果也非常丰富,人们合成了大量的生物活性多肽,包括催产素(oxytocin),胰岛素等,同时在多肽合成方法以及氨基酸保护基上面也取得了不少成绩,这为后来的固相合成方法的出现也提供了实验和理论基础。也就是这个阶段,Fred Sanger发明了氨基酸序列测定方法,并为此获得了1958年的Nobel化学奖。还是他后来发明了DNA序列检测方法,并于1980年再次获得了Nobel化学奖,成为到目前为止唯一获得两次Nobel化学奖的科学家。1963年,Merrifield提出了固相多肽合成方法(SPPS),这个在多肽化学上具有里程碑意义的合成方法,一出来,就由于其合成方便,迅速,现在已经成为多肽合成的首选方法,随后的发展也证明了该方法不仅仅是一种合成方法,而且也带来了有机合成上的一次革命,并成为了一支独立的学科,固相有机合成(SPOS)。当然,Merrifield也因此荣获了1984年的Nobel化学奖。也正是Merrifield,他经过了反复的筛选,最终屏弃了苄氧羰基(Z)在固相上的使用,首先将叔丁氧羰基(BOC)用于保护α-氨基并在固相多肽合成上使用,其可以在酸性条件下定量的脱除,反应也非常迅速,在30min就可以反应完全。由于叔丁氧羰基(BOC)方法中,氨基酸侧链的保护基团大多基于苄基(Bzl),因此也称为BOC-Bzl策略。同时,Merrifield在20世纪60年代末发明了第一台全自动多肽合成仪,并首次合成生物蛋白酶,核糖核酸酶(124个氨基酸)。随后的多肽化学研究主要集中在固相合成树脂,多肽缩合试剂,氨基酸保护基的研究。1972,Lou Carpino 首先将9-芴甲氧羰基(FMOC)用于保护α-氨基,其在碱性条件下可以迅速脱除,10min就可以反应完全,而且由于其反应条件温和,迅速得到广泛使用,到了20世纪80年代取代了叔丁氧羰基(BOC),成为了固相多肽合成中的首选合成方法。该方法中氨基酸的侧链大多基于叔丁基(But),因此,也称为FMOC-But策略。同时,在多肽合成树脂,缩合试剂以及氨基酸保护,包括合成环肽的氨基酸正交保护上也取得了丰硕的成果。
进入21世纪,随着蛋白质组学的研究深入,对于多肽化学的要求不仅仅是合成方法,而更多的集中在多肽标记与修饰方法,以及蛋白结构与功能模拟多肽的合成以及长肽或蛋白合成。
多肽化学合成的基本介绍
多肽化学合成方法,包括液相和固相两种方法。液相合成方法现在主要采用BOC和Z两种保护方法,现在主要应用在短肽合成,如阿斯巴甜,力肽,催产素等,其相对与固相合成,具有保护基选择多,成本低廉,合成规模容易放大的许多优点。与固相合成比较,液相合成主要缺点是,合成范围小,一般都集中在10个氨基酸以内的多肽合成,还有合成中需要对中间体进行提纯,时间长,工作量大。固相合成方法现在主要采用FMOC和BOC两种方法,它具有合成方便,迅速,容易实现自动化,而且可以比较容易的合成到30个氨基酸左右多肽。
1.1.氨基酸保护基
20种常见氨基酸,根据侧链可以分为几类:脂肪族氨基酸(Ala,Gly,Val,Leu,Ile,),芳香族氨基酸(Phe,Tyr,Trp,His),酰胺或羧基侧链氨基酸(Asp,Glu,Asn,Gln),碱性侧链氨基酸(Lys,Arg),含硫氨基酸(Cys,Met),含醇氨基酸(Ser,Thr),亚氨型基酸(Pro)。多肽化学合成中氨基酸的保护非常关键,直接决定了合成能够成功的关键。因为常见的20中氨基酸中有很多都是带有活性侧链的,需要进行保护,一般要求,这些保护基在合成过程中稳定,无副反应,合成结束后可以完全定量的脱除。合成中需要进行保护的氨基酸包括:Cys,Asp,Glu,His,Lys,Asn,Gln,Arg,Ser,Thr,Trp,Tyr。需要进行保护的基团:羟基,羧基,巯基,氨基,酰胺基,胍基,吲哚,咪唑等。其中Trp也可以不保护,因为吲哚性质比较稳定。当然在特殊的情况下,有些氨基酸也可以不保护,象,Asn,Gln ,Thr,Tyr。
表1 常见3种氨基脱除条件
|
|
TFA
|
HBr/TFA
|
H2/Pd-C
|
Piperidine/DMF
|
|
Boc
|
y
|
y
|
n
|
n
|
|
Z
|
n
|
y
|
y
|
n
|
|
Fmoc
|
n
|
y
|
n
|
y
|

(图1 常见3种氨基保护基结构)
氨基酸侧链保护基团非常多,同一个侧链有多种不同的保护基,可以在不同的条件下选择性的脱除,这点在环肽以及多肽修饰上具有很重要的意义。而且侧链保护基和选择的合成方法有密切的关系,液相和固相不一样,固相中BOC和FMOC策略也不一样,从某种意义上看,多肽化学就是氨基酸保护基的灵活运用与搭配。关于侧链保护基的使用,请参考王德心的《固相有机合成——原理及应用指南》第四章,我们这里主要介绍Cys,Lys,Asp的几种保护基及其脱除方法。Cys最常见的保护基有三种,Trt,Acm,Mob,这三个保护基可以完成多对二硫键多肽的合成。Lys最常见的保护基有:Boc,Fmoc,Trt,Dde,Allyl,这对于固相合成环肽提供了很多正交的保护策略。Asp最常见的保护基有:Otbu,OBzl,OMe,OAll,OFm,同样也提供了多种正交的保护策略。
表2 巯基常见保护基
|
简称
|
结构
|
脱除条件
|
|
Trt
|

|
TFA,HCl/HOAc,I2/MeOH
|
|
Acm
|

|
I2/MeOH,Hg2+
|
|
Mob
|

|
HF,TFMSA,Hg2+
|
(表3 氨基常见保护基)
|
简称
|
结构
|
脱除条件
|
|
Trt
|
|
TFA,HOAc,HCOOH
|
|
Boc
|

|
HCl/HOAc,TFA/DCM
|
|
Fmoc
|

|
Pip/DMF,NaOH/MeOH
|
|
Dde
|

|
H2NNH2/DMF
|
|
Allyl
|

|
Pd(Ph3P)4,吗啉/THF
|
(表4 羧基常见保护基)
|
简称
|
结构
|
脱除条件
|
|
Otbu
|

|
TFA,HOAc,HCOOH
|
|
OBzl
|

|
H2/Pd,HF,TFMSA
|
|
OMe
|

|
NaOH/MeOH
|
|
OAll
|
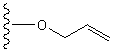
|
Pd(Ph3P)4,吗啉/THF
|
|
OFm
|

|
Pip/DMF,DBU/DMF
|
1.2.多肽缩合试剂
目前多肽合成中,主要采用羧基活化方法来完成接肽反应,最早使用的是将氨基酸活化为酰氯,叠氮,对称酸酐以及混合酸酐的方法,但是由于这些条件下,存在氨基酸消旋,以及反应试剂危险以及制备比较复杂,逐渐被后来的缩合试剂取代,按照其结构可以分为两种:缩合试剂主要有:碳二亚胺型,鎓盐型(Uronium)。
1.2.1.碳二亚胺型
主要包括:DCC,DIC,EDC.HCl等。采用DCC进行反应,由于反应中生成的DCU,在DMF中溶解度很小,产生白色沉淀,所以一般不用在固相合成中,但是由于其价格便宜,在液相合成中,可以通过过滤除去,应用仍然相当广泛。EDC.HCl因为其水溶解性的特点,在多肽与蛋白的连接中使用比较多,而且也相当成功。但是该类型的缩合试剂的一个最大的缺点,就是如果单独使用,会有比较多的副反应,但是研究表明如果在活化过程中添加HOBt,HOAt等试剂,可以将其副反应控制在很低的范围。其反应机理如下:

(图2 DIC活化反应机理)
1.2.2.鎓盐型
鎓盐型缩合试剂反应活性高,速度快,现在使用非常广泛,主要包括:HBTU,TBTU,HATU,PyBOP等。该试剂使用过程中需要添加有机碱,如,二异丙基乙胺(DIEA),N-甲基吗啉(NMM),该试剂加入后,才能活化氨基酸。其反应机理如下:

(图3 TBTU活化反应机理)
1.3.多肽合成方法比较
1.3.1.液相多肽合成(solution phase synthesis)
液相多肽合成现在仍然广泛的使用,在合成短肽和多肽片段上具有合成规模大,合成成本低的显著优点,而且由于是在均相中进行反应,可以选择的反应条件更加丰富,象一些催化氢化,碱性水解等条件,都可以使用,这在固相中,使用却由于反应效率低,以及副反应等原因,无法应用。液相多肽合成中主要采用BOC和Z两种反应策略。
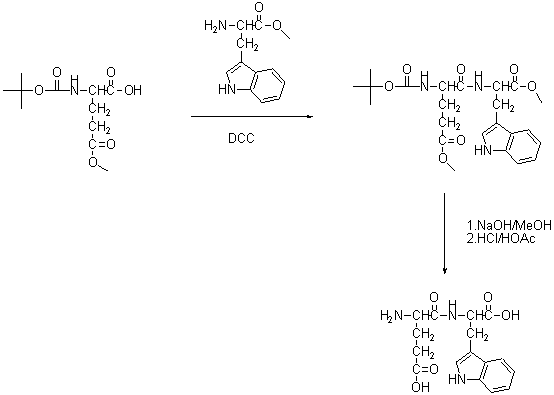
(图4液相合成Glu-Trp)
1.3.2.固相多肽合成

(图5 FMOC固相合成Glu-Trp)
固相多肽合成现在使用的主要有两种策略:BOC和FMOC两种。BOC方法合成过程中,需要反复使用TFA脱BOC,而且在最后从树脂上切割下来需要使用HF,由于HF必须使用专门的仪器进行操作,而且切割过程中容易产生副反应,因此现在使用受到实验条件限制,使用也逐渐减少。FMOC方法反应条件温和,在一般的实验条件下就可以进行合成,因此,也得到了非常广泛的应用。
1.3.2.1.固相合成中常用树脂
固相合成中树脂,一般都是聚苯乙烯-二乙烯苯材料,大小在75-150μm,交联度在1-2%之间,现在使用的大多是1%,因为这种交联度下,树脂在DMF,DCM中具有很好的溶胀性能,立体上是一个空间网状结构,反应物分子可以在树脂内部自由移动。树脂中最关键的部分是连接手臂,它一端连接在树脂上,一端作为反应位点。目前广泛使用的树脂有:PAM,MBHA,Wang,2-Cl-Trt,Rink-Amide-MBHA等。其中PAM,MBHA是用在BOC策略中,因为其对酸非常稳定,需要在HF,TFMSA等强酸条件下才能够切割下来。

(图6 固相合成常用树脂)
1.3.2.2.茚三酮检测
固相多肽合成中,主要是通过检测树脂上游离氨基来判断连接效率,检测方法称为Kaiser方法,其检测结果,如果有游离氨基的时候,显示兰色,或红褐色(pro,ser,His)。
Kaiser试剂包括:
A,6% 茚三酮的乙醇溶液
B,80% 苯酚的乙醇溶液
C,2% 0.001M KCN的吡啶溶液
配制中的吡啶需要经过茚三酮处理后,重蒸后再使用。检测过程,取少量树脂,加入A,B,C各2-3滴,100℃下加热1-2min,如果溶液有兰色,或树脂出现兰色,红褐色,表明还有游离氨基,否则说明连接完全。
还有其它检测游离氨基的方法:三硝基苯磺酸法,苦味酸法,溴芬兰法等。

(图7 茚三酮检测原理)
1.3.2.3.固相合成切割方法
固相合成完成之后,必须选择合适的切割试剂将多肽从树脂上切割下来,然后经过冰乙醚沉淀,离心收集沉淀,经过HPLC分离纯化,冷冻干燥得到最后产品。由于选择的树脂不同,氨基酸序列不同,在切割时候,选择的切割方法也不完全相同,一般都是选择酸性条件下切割的条件,对于PAM,MBHA树脂,一般采用HF切割,切割过程中需要添加对甲苯酚,对巯基苯酚,苯甲醚等试剂。而对于Wang,Rink-Amide,Trt树脂,一般采用TFA切割,切割过程中加入,乙二硫醇,苯甲硫醚,水,三异丙基硅烷,苯酚等。这些添加试剂主要作为碳正离子俘获试剂使用,目的是俘获切割反应过程中生成的碳正离子,减少这些碳正离子对部分氨基酸侧链的进攻导致的副反应,比较容易产生副反应的氨基酸有:Trp,Tyr。切割试剂用量一般10-15ml/g树脂。常用的切割配比:HF/p-cresol/p-thiocresol(90/5/5),TFA/TIS/EDT/H2O(94/1/2.5/2.5),反应一般是在室温条件下2h-4h。
1.3.3.多肽合成中主要问题
1.3.3.1.消旋及其反应机理
多肽合成过程中,部分氨基酸在活化的过程中会导致不同程度的消旋,特别容易消旋的氨基酸有:Cys,His,Phe,当然这些消旋化还和溶剂,温度以及合成中的有机碱等因素有关。对于这些氨基酸,可以通过采用高效缩合试剂,减少反应时间,可以减少消旋的比例,一般条件选择适当,消旋化都可以控制在5%以内。消旋反应机理如下:

(图8 消旋反应机理 )
1.3.3.2.二酮哌嗪(DKP)反应
DKP副反应出现在FMOC-Wang树脂合成过程中,主要出现在第一个氨基酸为Pro的时候,当第二个氨基酸脱FMOC的时候,α-氨基被游离出来之后,立即对Wang树脂的苄酯键进行分子内胺解,生成六元环二酮哌嗪衍生物,同时从Wang树脂上释放出来,导致反应终止。该反应非常迅速,文献报道采用50%Pip/DMF,脱1min,4min的条件,但是我们实验证明在多数情况下,即使是1min左右反应就超过了20%。因此一般在末端第一个氨基酸为Pro的时候,建议采用2-Cl-Trt树脂合成,由于该树脂巨大的空间阻力,可以完全消除该副反应。这个副反应在BOC策略合成过程中,却可以完全避免,因为BOC在使用TFA脱除后,使氨基以TFA盐的形式存在,从而失去了亲核性,不能进攻苄酯键。

(图9 DKP反应机理)
1.3.3.3.困难序列多肽合成
固相多肽合成中也经常遇到多肽合成失败或合成效率很低的问题,这里面的主要原因是由于多肽序列引起的,因为有些多肽序列在树脂上形成β-折叠,改变了树脂的溶胀性能,还有可能将反应的活性位点埋藏在树脂里面,这样使得反应很难进行,目前报道使用的主要方法有:
使用混合溶剂,DMSO/DMF,6N 胍啶/DMF溶液
提高反应温度,或采用微波方法
使用高离液盐,LiCl,NaClO4等。
使用溶胀性能更好的PEG-PS树脂,同时减少树脂担载量(0.05-0.2mmol/g)
1.4.合成多肽分析鉴定方法
多肽的分析鉴定方法有多肽一级结构,二级结构鉴定,多肽一级结构包括:质谱分析,氨基酸组成分析,氨基酸序列分析。二级结构包括:圆二色谱(CD),NMR,X-衍射等方法。
1.4.1.纯度分析
多肽的纯度分析,一般都采用HPLC进行分析,选择RP-C18,粒径5μm,孔径300A,4.6×150mm,流动相:A,0.1% TFA/H2O;B,0.1% TFA/ACN,洗脱梯度,5%B--65%B,时间30min。也有些多肽,特别是短肽,由于亲水性强,在C18上保留很弱,需要改变条件,这里主要有两种方法:一个改变分析梯度,可以将起始梯度改为2%,等度或小梯度洗脱;另外一个方法是在流动相中加入强离子对试剂,如七氟丁酸,十八烷基磺酸钠等。
1.4.2.一级结构分析
1.4.2.1.质谱分析
质谱分析的目的主要是确证分子量,当然采用MS/MS可以部分的了解多肽序列的信息,但是这个需要比较全的数据库作为基础,分析才能比较准确。由于多肽性质不稳定,需要采用软电离技术,目前多肽分析主要使用了电喷雾(ESI-MS),基质辅助激光解吸(MALDI-MS)。其中ESI-MS通常会给出多电荷峰,电荷数目和多肽序列上氨基,胍基,咪唑基的数目有关,因此,经常给出了双电荷,三电荷等离子峰。MALDI-MS可以分析蛋白大分子,而且通常情况下很少带多电荷,因此数据直接对应了分子离子峰,容易分析。此外,通常在分析长肽或蛋白过程中,需要添加NH4,Na,K等离子,提高灵敏度,所以一般在MS中出现一组峰,分别对应:(M+H)+,(M+NH4)+,(M+Na)+,(M+K)+。
1.4.2.2.氨基酸组成分析
氨基酸组成分析一般需要产品的纯度较高,它可以给出多肽中氨基酸的种类,数目。分析过程首先通过酸水解破坏肽键,典型酸水解的条件是:真空条件下,110℃,用6M盐酸水解16至72小时。酸水解虽然很有用,但酸水解条件下不能获得完整的氨基酸分析,因为天冬酰胺和谷氨酰胺的侧链含有酰胺键,用于切断蛋白质肽键的酸也可以将天冬酰胺转换为天冬氨酸,谷氨酰胺转换为谷氨酸。由于水解温度比较高,色氨酸的吲哚环容易被空气氧化,即使在密封的管中,色氨酸的吲哚环也几乎都被破坏了。因此蛋白质的色氨酸含量往往是通过它的紫外吸收光谱估计的,也可以通过碱水解分析色氨酸的含量。半胱氨酸在酸水解中也不能精确测定,要精确测量需要在蛋白质水解之前进行氧化或羧甲基化,形成的衍生物在酸水解之后才能定量。
1.4.2.3.氨基酸序列分析
氨基酸序列分析的基本原理是Edman降解,主要涉及耦联、水解、萃取和转换等4个过程。首先使用苯异硫氰酸酯(PITC)在pH9.0的碱性条件下对蛋白质或多肽进行处理,PITC与肽链的N-端的氨基酸残基反应,形成苯氨基硫甲酰(PTC)衍生物,即PTC-肽。然后PTC-肽用三氟乙酸处理,N-端氨基酸残基肽键被有选择地切断,释放出该氨基酸残基的噻唑啉酮苯胺衍生物。接下来将该衍生物用有机溶剂(例如氯丁烷)从反应液中萃取出来,而去掉了一个N-端氨基酸残基的肽仍留在溶液中。萃取出来的噻唑啉酮苯胺衍生物不稳定,经酸作用,再进一步环化,形成一个稳定的苯乙内酰硫脲(PTH)衍生物,即PTH-氨基酸。留在溶液中的减少了一个氨基酸残基的肽再重复进行上述反应过程,整个测序过程现在都是通过测序仪自动进行。
1.4.3.二级结构分析
1.4.3.1.圆二色谱(CD)
圆二色谱是一种特殊的吸收谱,它通过测量蛋白质等生物大分子的圆二色光谱,从而得到生物大分子的二级结构,简单、快捷,广泛应用在蛋白质折叠,蛋白质构象研究,酶动力学等领域。圆二色谱紫外区段(190-240nm),主要生色团是肽链,这一波长范围的CD谱包含了生物大分子主链构象的信息。α-螺旋构象的CD谱在222nm、208nm处呈负峰,在190nm附近有一正峰。β-折叠构象的CD谱,在217-218nm处有一负峰,在195-198nm处有一强的正峰。无规则卷曲构象的CD谱在198nm附近有一负峰,在220nm附近有一小而宽的正峰。
1.4.3.2.核磁共振(NMR)
随着二维、三维以及四维NMR的应用,分子生物学、计算机处理技术的发展,使NMR逐渐成为大分子结构物质分析的主要方法之一。NMR可用于确定氨基酸序列、分布以及构象。目前,NMR在分析分子中含少于30个氨基酸的小肽时是非常有用的,分析结果快速准确。
1.4.3.3.X-衍射
X-衍射可获得有关化合物晶型的直接信息,而且可以判断相对与绝对构型。
多肽标记及修饰
目前多肽标记及修饰的内容非常多,广泛应用在多肽药物,多肽生物学,多肽抗体以及多肽试剂的研究中。目前应用广泛的有:非放射性核素标记(C13,H2),荧光标记(FAM,FITC),生物素标记,磷酸化修饰等。
2.1.非放射性核素标记
目前在非放射性核素标记中,使用广泛的仍然是C13,H2,因为其使用安全,放射性小。现在有比较完全的非放射性标记的氨基酸,可以按照正常的多肽合成方法将标记好的氨基酸直接连接到多肽上。
2.2.荧光标记
荧光标记由于没有放射性,实验操作简单。因此,目前在生物学研究中荧光标记应用非常广泛,荧光标记方法与荧光试剂的结构有关系,对于有游离羧基的采用的方法与接肽反应相同,也采用HBTU/HOBt/DIEA方法连接。但是对于FITC标记,需要在连接FITC前,增加一个氨基己酸,避免在切割的过程中被TFA切割掉。
2.3.生物素标记
生物素-亲合素系统 (biotin-avidin system,BAS),是70年代后期应用于免疫学,并得到迅速发展的一种新型生物反应放大系统。由于它具有生物素与亲合素之间高度亲和力及多级放大效应,并与荧光素、酶、同位素等免疫标记技术有机地结合,使各种示踪免疫分析的特异性和灵敏度进一步提高。主要有用于标记多肽氨基的生物素N-羟基丁二酰亚胺酯(BNHS)和生物素对硝基酚酯(pBNP),其中以BNHS最常用,当然,也可以直接使用生物素也可以标记,因为其结构上有个游离的羧基,采用HBTU/HOBt/DIEA方法缩合,由于生物素的溶解度低,使用DMSO/DMF的混合溶剂增加溶解度。
2.4.磷酸肽合成
磷酸肽在生命过程中发挥重要作用,磷酸化的位置在多肽上的Ser,Thr,Tyr。目前磷酸肽合成一般都采用磷酸化氨基酸,目前使用的都是单苄基磷酸化氨基酸,Tyr也可以直接使用磷酸化氨基酸。磷酸化氨基酸的连接一般采用HBTU/HOBt/DIEA方法,但是目前采用该方法合成磷酸化也有缺点,特别是在合成多磷酸化多肽或长肽的时候,连接效率低,最后产品纯度很低,对于这种磷酸化多肽,我们考虑采用后磷酸化方法,其合成过程就是在多肽合成结束后,选择性脱去要标记的氨基酸的侧链保护基,对于Tyr,Thr可以直接使用侧链不保护的氨基酸进行反应,而Ser可以采用Fmoc-Ser(trt),在1% TFA/DCM条件下可以定量的脱除。后磷酸化,采用双苄基亚磷酰胺,四氮唑生成亚磷酰胺四唑活性中间体,连接到羟基上,随后在过氧酸下氧化生成磷酰基,完成反应。
蛋白结构与功能模拟多肽
多肽在与蛋白受体结合发挥功能的时候,总是先折叠出某些特殊的结构,多肽类似物合成主要是为了模拟这些结构,保持或提高生物活性,同时也为了改变多肽的稳定性,提高其抗酶解能力。
3.1.α-螺旋多肽
α-螺旋是蛋白结构中最为普通的一种,但是一般多肽在溶液中大多是无规卷曲的,目前使用最多的方法是多肽保持α-螺旋结构就是在多肽表面通过共价键将处在α-螺旋结构的两个氨基酸连接起来,选择的位点(i,i+4或i,i+7),选择的化学键包括:二硫键,硫醚键,酰胺键,烯烃键(RCM)等方法。
3.1.1.二硫键
二硫键广泛存在与蛋白结构中,对稳定蛋白结构具有非常重要的意义,二硫键一般是通过序列中的2个Cys的巯基,经氧化形成。形成二硫键的方法很多:空气氧化法,DMSO氧化法,过氧化氢氧化法等。二硫键的合成过程,可以通过Ellman检测以及HPLC检测方法对其反应进程进行监测。
3.1.2.硫醚键
硫醚键的形成可以通过序列中的Lys,将溴乙酸连接到Lys的侧链氨基上,利用其和巯基的特异性反应,反应在缓冲溶液中进行,迅速高效。
3.1.3.酰胺键
多肽的内酰胺环肽的合成一般是利用Lys,Asp(Glu)的选择性保护,在固相上直接环化。BOC策略中可以采用BOC-Lys(Fmoc),BOC-Asp(OFm),FMOC策略中可以采用FMOC-Lys(Aloc),FMOC-Asp(Allyl)。对于首尾环肽,还可以先合成保护的多肽,然后在液相中环化生成目标多肽。
3.1.4.烯烃键(RCM)
RCM反应是一个过渡金属催化反应,其反应中使用催化剂:(Cy3P)2Cl2Ru=CHPh,可以催化烯烃环化,过程中脱去一分子乙烯。
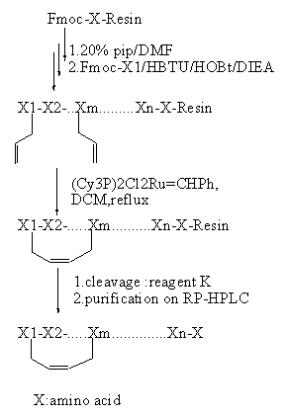
(图10 固相RCM反应 )
该反应也可以在固相树脂上直接环化,反应条件:15-25% (Cy3P)2Cl2Ru=CHPh DCM,60-80℃,24-48h。
3.2.TASP(template-assembled synthetic peptide)多肽
瑞士Basel大学的化学家首次提出TASP的概念,他们利用具有“发夹”结构的肽链作为模板,然后将具有α-螺旋结构的多肽直接连接到模板上,模拟蛋白质的高级结构。
3.3.长肽或蛋白合成
3.3.1.FMOC-tbu片段缩合方法
FMOC-tbu片段缩合方法在合成长肽以及蛋白上面运用非常广泛,其合成过程首先是合成保护性肽片段,经过纯化后,将几个片段在液相或固相上组装起来。使用本方法主要注意几个方面:片段的选择,片段的合成与纯化,片段缩合方法。片段选择要考虑到片段连接反应时间长,容易导致消旋,故片段的C末端最好选择Gly,Pro。合成的片段一般需要经过C4,C8等纯化,对于溶解性很差的多肽,可以采用硅胶柱层析方法进行纯化。由于片段的分子大,在树脂内移动速度慢,故反应时间一般都很长,而且反应过程中与片段浓度关系很大,溶解的时候,尽量提高片段的浓度。对多种条件的选择,发现采用DMF为溶剂,DIC/HOBt的方法缩合效率最高,消旋也最小。

(图11 FMOC-tbu片段缩合示意图)
3.3.2.自然化学连接(Native Chemical Ligation)
自然化学连接方法的优点是可以采用完全脱保护的多肽,因此不存在溶解性问题,其反应也是在缓冲水溶液中进行,由于其利用的是巯基和硫酯的特异性反应,再经过由S到N的转变完成肽键的合成。

(图13自然化学连接)
总之,今后多肽化学的研究,不仅是将更多的有机化学合成新方法,新技术引入到多肽研究当中,而且对蛋白的结构以及功能模拟,开展结构活性研究(structure activity relationship)将是研究开发的热点,因为这将为揭示蛋白的内在的生物活性本质提供大量实验数据。
|
多肽合成技巧
N末端的生物素标记
Wash 0.1 mmol resin with DMF.
Dissolve 0.244 g (+)-biotin (1 mmol, MW 244.3) in 5 mL DMF-DMSO (1:1) solution. A little warming is necessary.
Add 2.1 mL 0.45 M HBTU/HOBt solution and 0.3 mL DIEA to the solution prepared in step 2.
Add the activated biotin solution to the resin and let stir overnight.
Check resin to make sure coupling is complete as evidenced by negative ninhydrin test (colorless).
Wash resin with DMF-DMSO (1:1) (2x) to remove excess (+)-biotin.
Wash resin with DMF (2x) and DCM (2x).
Let the resin dry before proceeding to cleavage.
在2-氯TRT树脂上挂第一个FMOC氨基酸的方法
Weigh 10 g 2-chlorotrityl chloride resin (15 mmol) in a reaction vessel, wash with DMF (2x), swell the resin in 50 mL DMF for 10 min, drain vessel.
Weigh 10 mmol Fmoc-amino acid in a test tube, dissolve Fmoc-amino acid in 40 mL DMF, transfer the solution into the reaction vessel above, add 8.7 mL DIEA (50 mmol), swirl mixture for 30 min at room temperature.
Add 5 mL methanol into the reaction vessel and swirl for 5 min.
Drain and wash with DMF (5x).
Check substitution.
Add 50 mL 20% piperidine to remove the Fmoc group. Swirl mixture for 30 min.
Wash with DMF (5x), DCM (2x), put resin on tissue paper over a foam pad and let dry at room temperature overnight under the hood. Cover the resin with another piece of tissue paper, press lightly to break aggregates.
Weigh loaded resin.
Pack in appropriate container.
检测树脂上面是否挂上第一个FMOC氨基酸的方法
Weigh duplicate samples of 5 to 10 mg loaded resin in an eppendorf tube, add 1.00 mL 20% piperidine/DMF, shake for 20 min, centrifuge down the resin.
Transfer 100 µL of the above solution into a tube containing 10 mL DMF, mix well.
Pipette 2 mL DMF into each of the two cells (reference cell and sample cell), set spectrophotometer to zero. Empty the sample cell, transfer 2 mL of the solution from step 2 into the sample cell, check absorbance.
Subs = 101(A)/7.8(w)
A = absorbance
w = mg of resin
Check absorbance three times at 301 nm, calculate average substitution.
FMOC方法合成多肽操作步骤 (0.25 mmol)
Wash resin with DMF (4x) and then drain completely.
Add approximately 10 mL 20% piperidine/DMF to resin. Shake for one min and drain.
Add another 10 mL 20% piperidine/DMF. Shake for 30 min.
Drain reaction vessel and wash resin with DMF (4x). Make sure there is no piperidine remaining. Check beads using ninhydrin test, beads should be blue.
Coupling Step - Prepare the following solution:
1 mmol Fmoc-amino acid
2.1 mL 0.45 M HBTU/HOBT (1mmol)
348 µL DIEA (2 mmol)
Add above solution to the resin and shake for a minimum of 30 min. This coupling step can be longer if desired.
Drain reaction vessel and wash resin with DMF (4x).
Perform Ninhydrin test:
If negative (colorless), proceed to step 2 and continue synthesis.
If positive (blue), return to step 5 and re-couple the same Fmoc-amino acid. Increase the coupling time if necessary.